Oligo-RNA Combinatorial Assembly for the 3D modeling of RNA-protein complexes
The multiple roles of RNAs continue to be brought to light, and in particular those of a long time unsuspected high number of non-coding RNAs that are essential to cell physiology. Most of RNA’s roles are exerted in association with proteins, in a vast protein-RNA interactions network. The study of protein-RNA interactions is therefore of great importance in basic, clinical and pharmacological research. The structural description of a protein-RNA complex can make it possible to understand the recognition mechanisms and to regulate them. These structures are difficult to obtain experimentally, but can sometimes be modeled in silico, by docking. This technique consists in predicting the structure of a macro-molecular complex from a known structure of each of the partners.
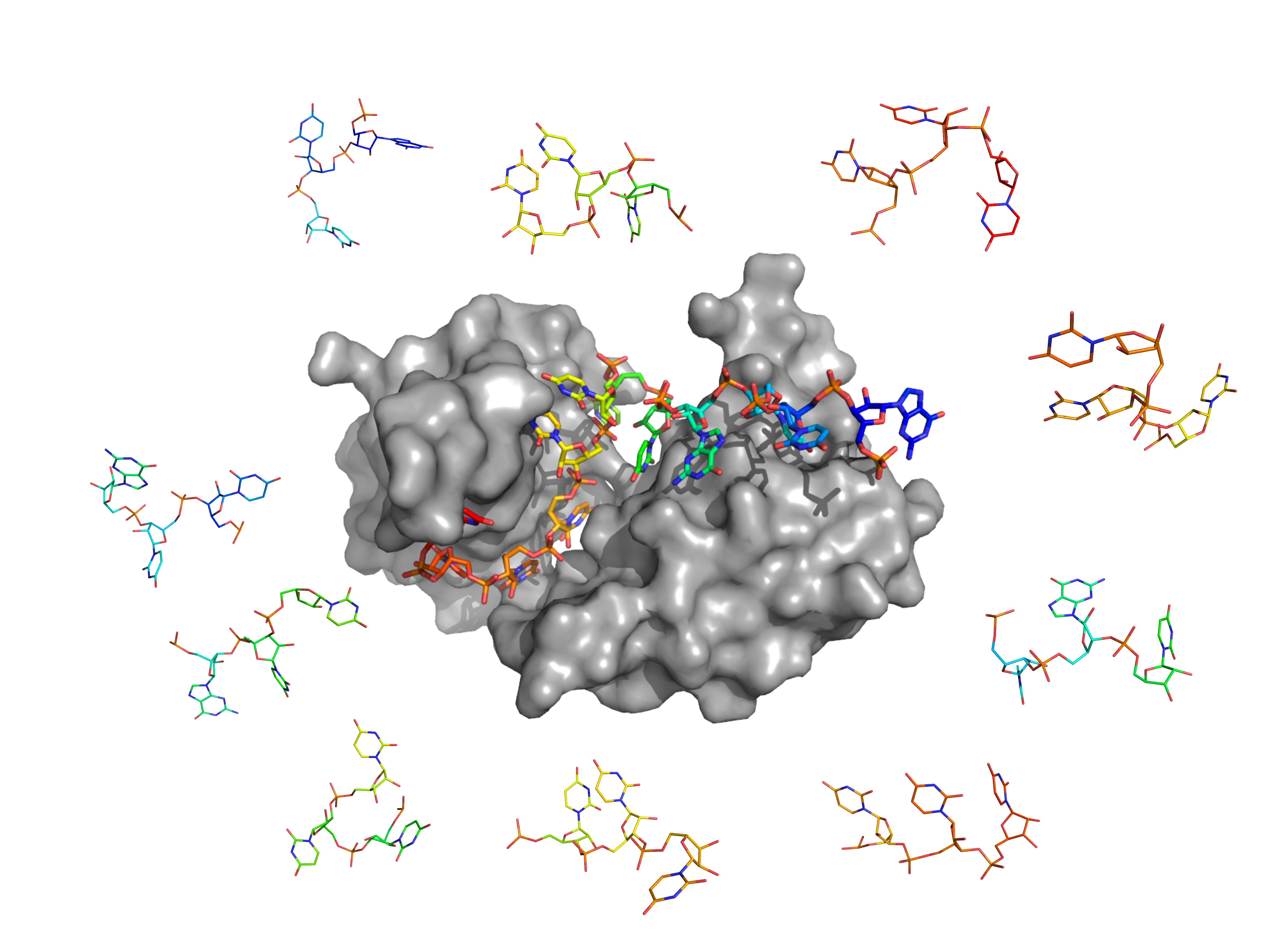
While AI-based approaches have made a tremendous progress in many docking applications (protein-protein, protein-ligand, etc), they are still impaired by the relatively low amount of protein-RNA data in regard to RNA conformational variability. Classical physics-based methods of protein-RNA docking are also severely limited by two difficulties: (i) They require a known structure of unrelated, often non-existent, RNA, and (ii) they can represent only one small part of the RNA flexibility. This is particularly problematic for hyper-flexible single-stranded RNA. These methods are therefore only efficient for a fraction of the structured RNAs, and can therefore not be generalized to large-scale studies. Especially, proteins containing RNA recognition motifs (“RRM” domains) represent >1% of the human proteom, and most bind single-stranded RNAs (ssRNAs). Other classes of proteins bind (partially) structured RNAs, i.e. containing double/triple stranded regions.
We seek to push back these limits, particularly through the development of fragment-based docking. We apply a double discretization of the ligand by (i) cutting it into fragments, and (ii) representing each fragment by an ensemble of rigid conformations. Those are docked onto the protein, then combinatorially reassembled into realistic full RNA models. In the case of RNA of unknown structure, this approach allows to bypasse a conformational exploration of the entire ligand, which is often extremely expensive in computing time, and rarely exhaustive in the case of a highly flexible linear polymer.
Publications:
A hybrid combinatorial method for docking ssRNA on proteins at the thermodynamic equilibrium , Chinmay Singhal, Yann Ponty, Isaure Chauvot de Beauchene. RECOMB 2018
Fragment-based modeling of protein-bound ssRNA , Isaure Chauvot de Beauchene, Sjoerd De Vries, Martin Zacharias. ECCB 2016
Fragment-based modelling of single stranded RNA bound to RNA recognition motif containing proteins , Isaure Chauvot de Beauchêne, Sjoerd De Vries, Martin Zacharias. Nucleic Acids Research, 2016
Binding Site Identification and Flexible Docking of Single Stranded RNA to Proteins Using a Fragment-Based Approach , Isaure Chauvot de Beauchêne, Sjoerd De Vries, Martin Zacharias. PLoS Computational Biology, 2016



